Page 1 of 1
CO3^{2-} Optimization
Posted: Wed Jun 01, 2022 2:32 am
by zhaoru_sun1
Dear all,
I am trying to optimize CO3^{2-} in gas. But I found it difficult to converge in electron step and ion step even with ALGO=ALL. Other species, such as CO2 and HCO3- converge more easily. So I think it might be because of the charges in CO3^{2-} ? Any suggestion wound be appreciated.
All relevant files are attached for reference.
Re: CO3^{2-} Optimization
Posted: Wed Jun 01, 2022 7:02 am
by martin.schlipf
Here are a couple of suggestions:
Start with the relaxations with a simpler system and increase the precision once you have some better initial guess. Specifically, run with a smaller cell, smaller cutoff, and PREC = Normal. Then converge these settings once you have a reasonable starting point.
Make sure you read the
section on dipole corrections, otherwise you may end up needing much larger cells than necessary.
If you set all magnetic moments to 0 it does not make sense to calculate spin polarized. With ISPIN = 1 the calculation should be more robust and faster.
Did you notice some issues with ALGO = Normal so that you switched to ALGO = All? We also recommend preconverging with the PBE functional if you struggle with SCAN.
Re: CO3^{2-} Optimization
Posted: Thu Jun 02, 2022 5:16 am
by zhaoru_sun1
martin.schlipf wrote: ↑Wed Jun 01, 2022 7:02 am
Start with the relaxations with a simpler system and increase the precision once you have some better initial guess. Specifically, run with a smaller cell, smaller cutoff, and PREC = Normal. Then converge these settings once you have a reasonable starting point.
The structure is simple, and the ion position change is less than 0.001 angstroms from XDATCAR. So I think the structure is really close to convergence.
I ignored the dipole correction before. But after adding IDIPOLE=4, the situation doesn't improve very well.
If you set all magnetic moments to 0 it does not make sense to calculate spin polarized. With ISPIN = 1 the calculation should be more robust and faster.
I'm not sure if the molecule has spin, the scf result shows it does, So I've changed INCAR to match the vasp result.
Did you notice some issues with ALGO = Normal so that you switched to ALGO = All? We also recommend preconverging with the PBE functional if you struggle with SCAN.
ALGO = Normal makes it harder for electrons convergence and dE fluctuates a lot. I learn about ALGO=ALL is more robust, so I use it.
Also, I noticed a strange thing. For the last two steps, the force is still greater than 0.01 even though there is no change in the atomic positions.
Code: Select all
POSITION TOTAL-FORCE (eV/Angst)
-----------------------------------------------------------------------------------
1.04442 0.72129 0.00000 -0.011212 -0.039821 -0.000000
0.09930 19.73300 -0.00000 -0.006612 0.000469 -0.000000
18.85591 0.54795 0.00000 -0.005073 -0.004393 -0.000000
0.00046 20.99776 0.00010 0.022898 0.043745 0.000000
-----------------------------------------------------------------------------------
total drift: -0.007120 0.034985 -0.098978
Code: Select all
POSITION TOTAL-FORCE (eV/Angst)
-----------------------------------------------------------------------------------
1.04442 0.72129 0.00000 -0.009083 -0.038509 -0.000000
0.09930 19.73300 -0.00000 -0.006426 -0.001790 -0.000000
18.85591 0.54795 0.00000 -0.007418 -0.003325 -0.000000
0.00046 20.99776 0.00010 0.022928 0.043624 0.000000
-----------------------------------------------------------------------------------
total drift: -0.006941 0.034736 -0.098989
New calculation files have been attached.
Re: CO3^{2-} Optimization
Posted: Thu Jun 02, 2022 8:17 am
by martin.schlipf
Did you stop the calculation with a STOPCAR file? It seems that the ionic relaxation is almost converged according to the criteria you specified, so one would not expect large changes in the positions anymore. If you look in the XDATCAR file or the xml file, you will see that there are still a very small change in the positions.
Regarding the IDIPOLE setting, that should not really help with the convergence of the electronic or ionic relaxation, but with converging the system with respect to the cell size.
I also looked at the electronic structure and it will be difficult to get this correct with DFT
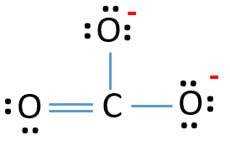
As you can see in the figure, the electronic state at the 3 oxygens is not the same. Instead two have single and one has a double bond. In reality which oxygen will have the double bond oscillates so in a mean-field theory like DFT you get a smeared-out state. As a result, you obtain a metallic solution.
What you can try is enforcing an insulating solution either with ISPIN = 2; NUPDOWN = 0 or ISMEAR = -2; FERWE = 12*1 4*0 and the releasing that constraint after the convergence to see if it stays in that solution.